对二代测序结果的下游分析软件很多,这里记录下使用 edgeR package 的使用方法。 edgeR 可以适用与 RNA-seq, SAGE-Seq 或者 ChIP-seq 数据的分析, degeR 基于 Robinson 和 Smyth 开发的精确统计方法来分析多 group 的实验结果,同时也基于 McCarthy 等人开发的广义线性模型(glms)方法来进行多因子实验的的统计分析。
edgeR 包的安装
- edgeR 包是基于 Bioconductor 平台发布的,所以安装不能直接用
install.packages()命令从 CRAN 上来下载 - 安装:
# try http:// if https:// URLs are not supported
>source("https://bioconductor.org/biocLite.R")
>biocLite("edgeR")
数据导入
- 由于 edgeR 对测序结果的下游分析是依赖 count 计数来进行基因差异表达分析的,在这里使用的是 featureCounts 来进行统计
.bam文件中 Map 的结果 - count 结果如下:
>library(edgeR)
>mydata <- read.table("counts.txt", header = TRUE, quote = '\t',skip =1)
>sampleNames <- c("CA_1","CA_2","CA_3","CC_1","CC_2","CC_3")
>names(mydata)[7:12] <- sampleNames
>head(mydata)
Geneid Chr Start End Strand Length CA_1 CA_2 CA_3 CC_1 CC_2 CC_3
1 gene1314 NW_139421.1 1257 1745 + 489 0 0 0 0 0 0
2 gene1315 NW_139421.1 2115 3452 + 1338 0 0 0 0 0 0
3 gene1316 NW_139421.1 3856 4680 + 825 0 0 0 0 0 0
4 gene1317 NW_139421.1 4866 5435 - 570 0 0 0 0 0 0
5 gene1318 NW_139421.1 6066 6836 - 771 0 0 0 0 0 0
6 gene1319 NW_139421.1 7294 9483 + 2190 0 0 0 0 0 0
- 在这里我们只是需要 Geneid 和后 6 列的样本的 count 信息来组成矩阵,所以要处理下
>countMatrix <- as.matrix(mydata[7:12])
>rownames(countMatrix) <-mydata$Geneid
>head(countMatrix)
CA_1 CA_2 CA_3 CC_1 CC_2 CC_3
gene1314 0 0 0 0 0 0
gene1315 0 0 0 0 0 0
gene1316 0 0 0 0 0 0
gene1317 0 0 0 0 0 0
gene1318 0 0 0 0 0 0
gene1319 0 0 0 0 0 0
要导入的矩阵由 3v3 样本组成(三组生物学重复)
创建 DEGlist
>group <- factor(c("CA","CA","CA","CC","CC","CC"))
>y <- DGEList(counts = countMatrix, group = group)
>y
An object of class "DGEList"
$counts
CA_1 CA_2 CA_3 CC_1 CC_2 CC_3
gene1314 0 0 0 0 0 0
gene1315 0 0 0 0 0 0
gene1316 0 0 0 0 0 0
gene1317 0 0 0 0 0 0
gene1318 0 0 0 0 0 0
14212 more rows ...
$samples
group lib.size norm.factors
CA_1 CA_1 1788537 1
CA_2 CA_2 1825546 1
CA_3 CA_3 1903017 1
CC_1 CC_1 1826042 1
CC_2 CC_2 2124468 1
CC_3 CC_3 2025063 1
过滤
- 过滤掉那些 count 结果都为 0 的数据,这些没有表达的基因对结果的分析没有用,过滤又两点好处:
-
可以减少内存的压力
-
可以减少计算的压力
>keep <- rowSums(cpm(y)>1) >= 2
>y <- y[keep, , keep.lib.sizes=FALSE]
>y
An object of class "DGEList"
$counts
CA_1 CA_2 CA_3 CC_1 CC_2 CC_3
gene1321 161 138 129 218 194 220
gene1322 2 3 1 1 3 3
gene1323 20 27 33 47 51 46
gene1324 60 87 79 86 100 132
gene1325 32 29 21 58 75 56
3877 more rows ...
$samples
group lib.size norm.factors
CA_1 CA_1 1788362 1
CA_2 CA_2 1825308 1
CA_3 CA_3 1902796 1
CC_1 CC_1 1825889 1
CC_2 CC_2 2124155 1
CC_3 CC_3 2024786 1
标准化处理
- edgeR采用的是 TMM 方法进行标准化处理,只有标准化处理后的数据才有可比性
>y <- calcNormFactors(y)
>y
An object of class "DGEList"
$counts
CA_1 CA_2 CA_3 CC_1 CC_2 CC_3
gene1321 161 138 129 218 194 220
gene1322 2 3 1 1 3 3
gene1323 20 27 33 47 51 46
gene1324 60 87 79 86 100 132
gene1325 32 29 21 58 75 56
3877 more rows ...
$samples
group lib.size norm.factors
CA_1 CA_1 1788362 0.9553769
CA_2 CA_2 1825308 0.9052539
CA_3 CA_3 1902796 0.9686232
CC_1 CC_1 1825889 0.9923455
CC_2 CC_2 2124155 1.1275178
CC_3 CC_3 2024786 1.0668754
设计矩阵
- 为什么要一个设计矩阵呢,道理很简单,有了一个设计矩阵才能够更好的分组分析
>subGroup <- factor(substring(colnames(countMatrix),4,4))
>design <- model.matrix(~ subGroup+group)
>rownames(design) <- colnames(y)
>design
(Intercept) subGroup2 subGroup3 groupCC
CA_1 1 0 0 0
CA_2 1 1 0 0
CA_3 1 0 1 0
CC_1 1 0 0 1
CC_2 1 1 0 1
CC_3 1 0 1 1
attr(,"assign")
[1] 0 1 1 2
attr(,"contrasts")
attr(,"contrasts")$subGroup
[1] "contr.treatment"
attr(,"contrasts")$group
[1] "contr.treatment"
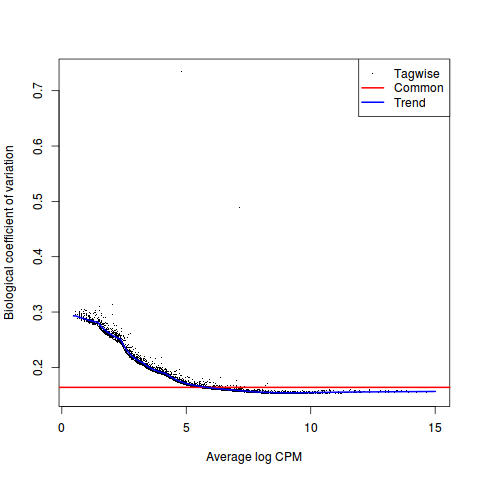
评估离散度
>y <- estimateDisp(y, design, robust=TRUE)
>y$common.dispersion
[1] 0.02683622
#plot
>plotBCV(y)
差异表达基因
> fit <- glmQLFit(y, design, robust=TRUE)
> qlf <- glmQLFTest(fit)
> topTags(qlf)
Coefficient: groupCC
logFC logCPM F PValue FDR
gene7024 -5.515648 9.612809 594.9232 6.431484e-44 2.496702e-40
gene6612 5.130282 8.451143 468.2060 1.557517e-39 3.023140e-36
gene2743 4.377492 5.586773 208.0268 3.488383e-26 4.513967e-23
gene12032 4.734383 5.098148 192.9378 4.359649e-25 4.231040e-22
gene491 -2.733910 10.412673 190.9839 6.104188e-25 4.739291e-22
gene8941 2.997185 6.839106 177.7614 6.332836e-24 4.097345e-21
gene2611 -2.846924 7.216173 174.7332 1.099339e-23 6.096619e-21
gene6242 2.529125 9.897771 169.2658 3.022914e-23 1.466869e-20
gene7252 3.732315 6.137670 188.2094 3.890569e-23 1.678132e-20
gene6125 2.875423 6.569935 160.3189 1.656083e-22 6.428914e-20
查看差异表达基因原始的 CMP
> top <- rownames(topTags(qlf))
> cpm(y)[top,]
CA_1 CA_2 CA_3 CC_1 CC_2 CC_3
gene7024 1711.383002 1405.861899 1480.121115 33.11418 37.16040 29.62696
gene6612 17.558649 12.103848 26.585753 403.99298 582.45796 1044.35046
gene2743 4.682306 1.815577 5.968230 62.91694 87.26431 114.34156
gene12032 1.755865 2.420770 2.712832 65.67646 47.59872 75.45617
gene491 2811.139727 2059.469669 2222.351938 444.83381 385.38258 253.68087
gene8941 23.996820 24.812888 24.415488 131.35291 244.67410 225.90560
gene2611 245.821088 310.463691 225.165052 43.04843 26.30455 39.81123
gene6242 231.188880 299.570228 298.411515 1348.29899 1343.61988 2191.93237
gene7252 9.364613 13.314232 5.425664 92.71970 108.55847 181.92807
gene6125 23.411532 14.524617 29.841152 145.70239 160.75005 185.16852
查看上调和下调基因的数目
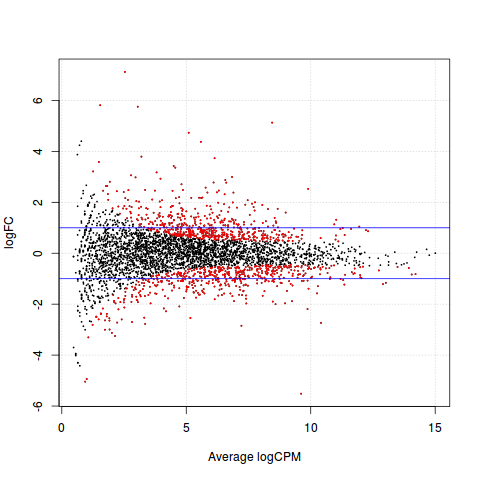
> summary(dt <- decideTestsDGE(qlf))
[,1]
-1 536
0 2793
1 553
挑选出差异表达基因的名字
> isDE <- as.logical(dt)
> DEnames <- rownames(y)[isDE]
> head(DEnames)
[1] "gene1325" "gene1326" "gene1327" "gene1331" "gene1340" "gene1343"
差异表达基因画图
>plotSmear(qlf, de.tags=DEnames)
>abline(h=c(-1,1), col="blue")